Alterations in the phosphoinositide-3-kinase/Akt/mammalian target of rapamyc in signaling pathway have been found in many human tumors. In particular, amplification and mutation of PIK3CA, mutation of PIK3R and Akt, and loss of tumor suppressor PTENcontribute to constitutive activation of this signaling pathway. Understanding the interplay among signaling molecules in the PI3K/Akt/mTOR pathway is of utmost importance. Two distinct mTOR complexes, mTORC1 and mTORC2, have been identified and have differential sensitivity to rapamycin. mTORC1 is downstream of Akt, sensitive to rapamycin inhibition, and controls cap-dependent protein translation. The two best-studied mTORC1 substrates are 40S ribosomal S6 LY294002 154447-36-6 kinase 1and eukaryotic translation initiation factor 4E-binding protein 1, which mediate efficient protein translation. In contrast, mTORC2 is directly upstream of Akt and is resistant to rapamycin. Akt can be activated by phosphorylation at two different 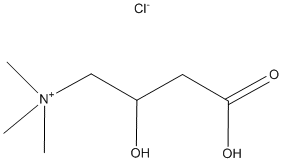 sites, S473 by mTORC2 and T308 by phosphoinositide-dependent kinase 1. Constitutive activation of the PI3K/Akt/mTOR signaling axis leads to uncontrolled tumor cell proliferation and survival. Given the importance of the mTOR pathway in cancer cell growth, significant efforts have attempted to identify targeted inhibitors. Rapamycin and its analogs, such as RAD001, AP23573and CCI-779are allosteric inhibitors of mTOR. The limited anticancer efficacy of the rapalogs can be explained by two possible mechanisms:rapalogs inhibit only mTORC1, thereby inducing feedback activation of survival signaling pathways such as Akt phosphorylation; orrapalogs incompletely block mTORC1 downstream signaling. For example, in some cells mTOR inhibitors prevent phosphorylation of S6K1 but not 4E-BP1, thus allowing the cells to escape growth inhibition. Previous studies indicate that PTEN inactivation, PIK3CA mutation, and mTOR dysregulation are common molecular signatures for endometrial carcinoma. Furthermore, PI3K activation is a hallmark for aggressive tumors at this site. mTOR inhibitorshave been tested in phase I and II clinical trials for advanced and recurrent endometrial carcinomas with some promising clinical outcomes; however, response rates are not robust. In general, responses are partial and vary from 8%�C26% with an additional 20%�C63% of patients achieving stable disease for at least four months. Some patients achieve no benefit from therapy, whereas in others, stable disease or an initial response occurs. Nevertheless, most patients eventually experience progression of disease. More information will be available following the analysis of the phase II trial of Adriamycin temsirolimus for advanced endometrial cancer, Gynecologic Oncology Group trial 248; however, since this trial only recently closed to accrual, the outcome data are not mature. In this current study, we investigated how inhibition of mTOR can be optimized. We examined the growth inhibitory effect of temsirolimus on a panel of endometrial cancer cells and observed differential sensitivity as well as compensatory Akt phosphorylation in a subset of cell lines, which may represent one mechanism for acquired resistance. We identified cells which were primarily resistant to treatment and compared these to other cells which initially responded but employed escape mechanisms to achieve acquired resistance. To overcome both forms of resistance, we applied dual inhibition of PI3K and mTOR to prevent cell survival signaling. Our data reveal that combination treatment of temsirolimus with either BEZ235, a dual PI3K/mTOR inhibitor, or ZSTK474, a pan PI3K inhibitor, blocked Akt activation and inhibited phosphorylation of both 4E-BP1 and the substrate for S6K, ribosomal S6, which ultimately resulted in synergistic.
sites, S473 by mTORC2 and T308 by phosphoinositide-dependent kinase 1. Constitutive activation of the PI3K/Akt/mTOR signaling axis leads to uncontrolled tumor cell proliferation and survival. Given the importance of the mTOR pathway in cancer cell growth, significant efforts have attempted to identify targeted inhibitors. Rapamycin and its analogs, such as RAD001, AP23573and CCI-779are allosteric inhibitors of mTOR. The limited anticancer efficacy of the rapalogs can be explained by two possible mechanisms:rapalogs inhibit only mTORC1, thereby inducing feedback activation of survival signaling pathways such as Akt phosphorylation; orrapalogs incompletely block mTORC1 downstream signaling. For example, in some cells mTOR inhibitors prevent phosphorylation of S6K1 but not 4E-BP1, thus allowing the cells to escape growth inhibition. Previous studies indicate that PTEN inactivation, PIK3CA mutation, and mTOR dysregulation are common molecular signatures for endometrial carcinoma. Furthermore, PI3K activation is a hallmark for aggressive tumors at this site. mTOR inhibitorshave been tested in phase I and II clinical trials for advanced and recurrent endometrial carcinomas with some promising clinical outcomes; however, response rates are not robust. In general, responses are partial and vary from 8%�C26% with an additional 20%�C63% of patients achieving stable disease for at least four months. Some patients achieve no benefit from therapy, whereas in others, stable disease or an initial response occurs. Nevertheless, most patients eventually experience progression of disease. More information will be available following the analysis of the phase II trial of Adriamycin temsirolimus for advanced endometrial cancer, Gynecologic Oncology Group trial 248; however, since this trial only recently closed to accrual, the outcome data are not mature. In this current study, we investigated how inhibition of mTOR can be optimized. We examined the growth inhibitory effect of temsirolimus on a panel of endometrial cancer cells and observed differential sensitivity as well as compensatory Akt phosphorylation in a subset of cell lines, which may represent one mechanism for acquired resistance. We identified cells which were primarily resistant to treatment and compared these to other cells which initially responded but employed escape mechanisms to achieve acquired resistance. To overcome both forms of resistance, we applied dual inhibition of PI3K and mTOR to prevent cell survival signaling. Our data reveal that combination treatment of temsirolimus with either BEZ235, a dual PI3K/mTOR inhibitor, or ZSTK474, a pan PI3K inhibitor, blocked Akt activation and inhibited phosphorylation of both 4E-BP1 and the substrate for S6K, ribosomal S6, which ultimately resulted in synergistic.