The proprotein convertases are a Niraparib in vivo family of nine serine proteases implicated in the processing of a multitude of precursor proteins. The first seven members activate a large number of polypeptide hormones, growth factors, adhesion molecules, various viral surface proteins and protoxins of bacteria by cleavage at basic residues. The eighth and ninth members do not require a basic residue for cleavage and they play major roles in regulation of lipid homeostasis. Accumulated evidence over the last decade has confirmed PCs as potential therapeutic targets for several important pathologies including osteoarthritis, cancer, cardiovascular disease and viral infections. Therefore, development of PC MLN4924 inhibitors is clearly an important research and development field. Our interest in PC inhibitors originated from studies aiming at inhibiting PC6 in the female reproductive tract to inhibit embryo implantation. Uterine PC6 is pivotal in embryo implantation and is essential for the establishment of pregnancy. To enable implantation, the uterus must acquire epithelial receptivity and undergo a process known as decidualization to differentiate stromal fibroblasts into phenotypically and functionally distinct decidual cells. We have previously shown that PC6 is critical for both uterine epithelial receptivity and stromal cell decidualization. Knockdown of PC6 in a human endometrial epithelial cell line HEC1A significantly reduced its receptivity for blastocyst adhesion. Decidualization of primary human endometrial stromal cells was inhibited when PC6 activity was blocked. It has also been demonstrated in mice that when uterine PC6 production was blocked, decidualization was inhibited and implantation was prevented. In addition, PCs including PC6 also play an important role in HIV infection. Therefore, inhibition of PC6 is an attractive approach to develop novel, non-hormonal and female-controlled contraceptives that could also protect women from HIV infection. The majority of PC inhibitors reported in the literature to date have been proteins or peptides. Nona-D-arginine is one of the most potent peptide based PC inhibitors known to date. Poly R inhibits PC6 in vitro with a Ki in the nanomolar range and has been shown to inhibit HIV in cell culture. We have previously demonstrated that Poly R inhibits decidualization of HESC in culture and have evaluated the therapeutic potential of a PEGylated Poly R in inhibition of implantation in rabbits. However, the physiochemical properties of Poly R could limit their usefulness in therapeutic applications in women. Therefore, we continue to search for potent PC6 inhibitors with the desired characteristics such as serum stability and cell permeability. In this study, we evaluated five synthetic small molecule compounds derived from 2,5-dideoxystreptamine chemical scaffold previously reported by Jiao et al., 2006. Four of these compounds were previously shown to be potent inhibitors of both human furin and PC6 in vitro. Compound 1o was shown to be a relatively poor inhibitor of furin but no data on PC6 was reported. Here, the inhibitory potency of all five compounds against human PC6 was determined in vitro. In silico docking studies were performed to visualise the potential binding mode of these inhibitors in the active site of hPC6 and to gain an understanding of how this may relate to their inhibitory activity. The therapeutic potential of these small molecule inhibitors was then examined in in vitro human cell-based models to investigate their ability to inhibit two important PC6-mediated cellular processes essential for embryo implantation: decidualization of primary HESCs and attachment of human trophoblast spheroids to endometrial epithelial cells. PC6 plays a crucial role in embryo implantation and HIV infection; it is therefore highly desirable to develop inhibitors of PC6 for potential non-hormonal female contraceptives that could also protect women from HIV. In the ongoing search for PC6 inhibitors with appropriate physiochemical characteristics for therapeutic applications, we investigated 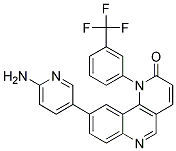 five synthetic small molecule compounds that had been previously reported as inhibitors of furin, another PC member. Our studies revealed that all five compounds were potent inhibitors against rhPC6 in vitro and they were able to adopt similar binding modes in the hPC6 active site. However, the functional studies by in vitro cell-based model demonstrated that only compound 1o was able to inhibit decidualization of HESCs.
five synthetic small molecule compounds that had been previously reported as inhibitors of furin, another PC member. Our studies revealed that all five compounds were potent inhibitors against rhPC6 in vitro and they were able to adopt similar binding modes in the hPC6 active site. However, the functional studies by in vitro cell-based model demonstrated that only compound 1o was able to inhibit decidualization of HESCs.
Monthly Archives: July 2019
in the left half of oriC and flanking the DUE region is essential for helicase loading is stimulated by the formation of DnaA sub-complex
At initiation DnaA-ATP molecules cooperatively bind the left half of the origin to form a right-handed DnaA-ATP helix, where individual DnaA molecules interact through their AAA+ domains, with oriC DNA wrapped around it. Binding of IHF immediately upstream of the DUE flanking R1 DnaA-box introduces a 160u bend in the DNA reversing the orientation of the DNA helical axis and assist in melting the DUE region. One of the exposed single-stranded DUE regions is fixed by binding the existing DnaA-ATP helix while the other strand is exposed for DnaC assisted DnaB helicase loading by the DnaA molecule bound to the R1 box. Further opening of the duplex allows for loading of the second helicase by one or more N-terminal domains of the DnaA-ATP filament. Although SJN 2511 promoted by formation of a DnaA oligomer on oriC, the exact mechanism for helicase loading at the origin differ between bacteria. After helicase loading, a cascade of events leading to replisome assembly and the beginning of the elongation follows. The replisome structure was recently covered in an excellent review and consists of a primosome complex and a PolIII holoenzyme complex, where each PolIII holoenzyme complex can be further divided into three different complexes: PolIII core, the sliding clamp and the clamp loader. The core polymerase needs the sliding clamp for processivity, which in turn is loaded onto the DNA by the clamp loader. In the firmicutes including S. aureus, the process of elongation is similar to that in E. coli with a couple of notable exceptions. The S. aureus helicase is loaded by the DnaI helicase loader assisted by the DnaB and DnaD proteins and two different replicative polymerases are used. The DnaE which is homologous to the E. coli PolIIIa only extends RNA AZ 960 primers initially and hands them off to PolC which is responsible for the processive synthesis. A third difference was recently revealed. Primer hand off in Bacillus subtilis, can occur after the synthesis of only two nucleotides by the DnaG primase and does not require other replication proteins. This is in contrast to the three-point switch hand off mechanism in E. coli. Here the x polypeptide of the clamp loader interacts with SSB to displace DnaG from the SSB-DnaG complex resulting in release of the primer which is then extended by the processive polymerase. In all bacteria examined so far the ring shaped b-clamp is a homodimer which encircles the DNA and slides along the duplex bringing the polymerase into contact with the DNA to ensure processivity. The b-clamp interacts with many different proteins including DnaE, PolC, d, PolIV, PolV, PolI, MutS, MutL, DNA ligase and Hda. These proteins all contain a conserved b-binding motif which binds a hydrophobic pocket located in each DnaN protomer. The b-sliding clamp has been the target  for potential new antibiotics and two different approaches have been used to identify compounds that block the peptide-binding pocket of b. First, synthetic peptides containing the beta-binding domain QLD/SLF were found to inhibit PolC-b2 and d-b2 interactions and similarly peptides containing b-binding sequence from d and Hda bound the b-clamp and inhibited DNA synthesis in vitro. Subsequently more efficient binders were identified by modification of the b-binding domain and these optimized peptide motifs have served as starting point for small molecule mimics to identify compounds that inhibit the a-b2 interaction at micromolar concentrations. In the second approach, a fluorescence based peptide displacement assay was used to identify small compounds that bind to the peptide-binding pocket of b. One compound, RU7, which inhibited PolII, PolIII and PolIV although to different extents was identified from a collection of 30,600 polar organic compounds. It was suggested that RU7 can be used as a starting point for rational drug design to create stronger inhibitors of replication. A fairly unexploited class of compounds that has attracted attention as putative antimicrobials is peptides. The extensively studied natural antimicrobial peptides are produced by multicellular organisms and the majority act by insertion and alteration/ damage of cytoplasmic membranes via formation of ion channels or transmembrane pores, but other have been associated with intracellular targets such as DNA and RNA synthesis and inhibition of enzymatic activities. This indicates that certain peptides can traverse the bacterial membrane to find their intracellular targets.
for potential new antibiotics and two different approaches have been used to identify compounds that block the peptide-binding pocket of b. First, synthetic peptides containing the beta-binding domain QLD/SLF were found to inhibit PolC-b2 and d-b2 interactions and similarly peptides containing b-binding sequence from d and Hda bound the b-clamp and inhibited DNA synthesis in vitro. Subsequently more efficient binders were identified by modification of the b-binding domain and these optimized peptide motifs have served as starting point for small molecule mimics to identify compounds that inhibit the a-b2 interaction at micromolar concentrations. In the second approach, a fluorescence based peptide displacement assay was used to identify small compounds that bind to the peptide-binding pocket of b. One compound, RU7, which inhibited PolII, PolIII and PolIV although to different extents was identified from a collection of 30,600 polar organic compounds. It was suggested that RU7 can be used as a starting point for rational drug design to create stronger inhibitors of replication. A fairly unexploited class of compounds that has attracted attention as putative antimicrobials is peptides. The extensively studied natural antimicrobial peptides are produced by multicellular organisms and the majority act by insertion and alteration/ damage of cytoplasmic membranes via formation of ion channels or transmembrane pores, but other have been associated with intracellular targets such as DNA and RNA synthesis and inhibition of enzymatic activities. This indicates that certain peptides can traverse the bacterial membrane to find their intracellular targets.
Single agent rapalogs have only achieved modest antitumor activity in the clinic
Alterations in the phosphoinositide-3-kinase/Akt/mammalian target of rapamyc in signaling pathway have been found in many human tumors. In particular, amplification and mutation of PIK3CA, mutation of PIK3R and Akt, and loss of tumor suppressor PTENcontribute to constitutive activation of this signaling pathway. Understanding the interplay among signaling molecules in the PI3K/Akt/mTOR pathway is of utmost importance. Two distinct mTOR complexes, mTORC1 and mTORC2, have been identified and have differential sensitivity to rapamycin. mTORC1 is downstream of Akt, sensitive to rapamycin inhibition, and controls cap-dependent protein translation. The two best-studied mTORC1 substrates are 40S ribosomal S6 LY294002 154447-36-6 kinase 1and eukaryotic translation initiation factor 4E-binding protein 1, which mediate efficient protein translation. In contrast, mTORC2 is directly upstream of Akt and is resistant to rapamycin. Akt can be activated by phosphorylation at two different  sites, S473 by mTORC2 and T308 by phosphoinositide-dependent kinase 1. Constitutive activation of the PI3K/Akt/mTOR signaling axis leads to uncontrolled tumor cell proliferation and survival. Given the importance of the mTOR pathway in cancer cell growth, significant efforts have attempted to identify targeted inhibitors. Rapamycin and its analogs, such as RAD001, AP23573and CCI-779are allosteric inhibitors of mTOR. The limited anticancer efficacy of the rapalogs can be explained by two possible mechanisms:rapalogs inhibit only mTORC1, thereby inducing feedback activation of survival signaling pathways such as Akt phosphorylation; orrapalogs incompletely block mTORC1 downstream signaling. For example, in some cells mTOR inhibitors prevent phosphorylation of S6K1 but not 4E-BP1, thus allowing the cells to escape growth inhibition. Previous studies indicate that PTEN inactivation, PIK3CA mutation, and mTOR dysregulation are common molecular signatures for endometrial carcinoma. Furthermore, PI3K activation is a hallmark for aggressive tumors at this site. mTOR inhibitorshave been tested in phase I and II clinical trials for advanced and recurrent endometrial carcinomas with some promising clinical outcomes; however, response rates are not robust. In general, responses are partial and vary from 8%�C26% with an additional 20%�C63% of patients achieving stable disease for at least four months. Some patients achieve no benefit from therapy, whereas in others, stable disease or an initial response occurs. Nevertheless, most patients eventually experience progression of disease. More information will be available following the analysis of the phase II trial of Adriamycin temsirolimus for advanced endometrial cancer, Gynecologic Oncology Group trial 248; however, since this trial only recently closed to accrual, the outcome data are not mature. In this current study, we investigated how inhibition of mTOR can be optimized. We examined the growth inhibitory effect of temsirolimus on a panel of endometrial cancer cells and observed differential sensitivity as well as compensatory Akt phosphorylation in a subset of cell lines, which may represent one mechanism for acquired resistance. We identified cells which were primarily resistant to treatment and compared these to other cells which initially responded but employed escape mechanisms to achieve acquired resistance. To overcome both forms of resistance, we applied dual inhibition of PI3K and mTOR to prevent cell survival signaling. Our data reveal that combination treatment of temsirolimus with either BEZ235, a dual PI3K/mTOR inhibitor, or ZSTK474, a pan PI3K inhibitor, blocked Akt activation and inhibited phosphorylation of both 4E-BP1 and the substrate for S6K, ribosomal S6, which ultimately resulted in synergistic.
sites, S473 by mTORC2 and T308 by phosphoinositide-dependent kinase 1. Constitutive activation of the PI3K/Akt/mTOR signaling axis leads to uncontrolled tumor cell proliferation and survival. Given the importance of the mTOR pathway in cancer cell growth, significant efforts have attempted to identify targeted inhibitors. Rapamycin and its analogs, such as RAD001, AP23573and CCI-779are allosteric inhibitors of mTOR. The limited anticancer efficacy of the rapalogs can be explained by two possible mechanisms:rapalogs inhibit only mTORC1, thereby inducing feedback activation of survival signaling pathways such as Akt phosphorylation; orrapalogs incompletely block mTORC1 downstream signaling. For example, in some cells mTOR inhibitors prevent phosphorylation of S6K1 but not 4E-BP1, thus allowing the cells to escape growth inhibition. Previous studies indicate that PTEN inactivation, PIK3CA mutation, and mTOR dysregulation are common molecular signatures for endometrial carcinoma. Furthermore, PI3K activation is a hallmark for aggressive tumors at this site. mTOR inhibitorshave been tested in phase I and II clinical trials for advanced and recurrent endometrial carcinomas with some promising clinical outcomes; however, response rates are not robust. In general, responses are partial and vary from 8%�C26% with an additional 20%�C63% of patients achieving stable disease for at least four months. Some patients achieve no benefit from therapy, whereas in others, stable disease or an initial response occurs. Nevertheless, most patients eventually experience progression of disease. More information will be available following the analysis of the phase II trial of Adriamycin temsirolimus for advanced endometrial cancer, Gynecologic Oncology Group trial 248; however, since this trial only recently closed to accrual, the outcome data are not mature. In this current study, we investigated how inhibition of mTOR can be optimized. We examined the growth inhibitory effect of temsirolimus on a panel of endometrial cancer cells and observed differential sensitivity as well as compensatory Akt phosphorylation in a subset of cell lines, which may represent one mechanism for acquired resistance. We identified cells which were primarily resistant to treatment and compared these to other cells which initially responded but employed escape mechanisms to achieve acquired resistance. To overcome both forms of resistance, we applied dual inhibition of PI3K and mTOR to prevent cell survival signaling. Our data reveal that combination treatment of temsirolimus with either BEZ235, a dual PI3K/mTOR inhibitor, or ZSTK474, a pan PI3K inhibitor, blocked Akt activation and inhibited phosphorylation of both 4E-BP1 and the substrate for S6K, ribosomal S6, which ultimately resulted in synergistic.
The CRLs compared to a more upstream enzyme would have the potential to only stabilize a particular subset of proteins
Possibly resulting in an improved selectivity profile. The NAE inhibitor MLN4924 was recently reported to be effective against both solidand hematologicalhuman cancer cells. We have previously employed high-throughput virtual screening to identify 6,699-biapigenin as only the second inhibitor of NEDD8-activating enzyme from a natural product and natural product-like database. While transition metal complexes have been widely utilized for the treatment of cancer, their SCH772984 activity against NEDD8-activating enzyme has not been explored. Inspired by the above findings as well as pioneering works from the Meggers’s group on the design of structurally rigid octahedral rutheniumand iridiumcomplexes as shape-complementary inhibitors of protein kinases, we sought to investigate the biological effects of a series of cyclometallated rhodium complexes on the NEDD8 pathway. Cyclometallated rhodium complexes containing the dipyridophenazine dipyridophenazinescaffold were chosen because of the following reasons: 1) the rhodium complex adopts an octahedral geometry rather than a square planar or tetrahedral symmetry, thus allowing much larger structural complexity for potential use in drug design; 2) the octahedral geometry of the rhodium complex provides a globular and rigid scaffold with limited conformational freedoms of the coligands that may interact with the previously inaccessible regions of chemical space in NAE; 3) the synthetic route for 1 is modular and convenient, thus allowing structural modification without the need for lengthy synthetic protocols; and 4) the extended aromatic dppz ligand structurally resembles the planar nature of NAE inhibitor 6,699-biapigenin, potentially functioning as the recognition arm for NAE. We report herein the synthesis and characterization of the racemic mixture of rhodium complex+and its analogues. Complex 1 was found to inhibit NAE activity in vitro and in cellulo. We then investigated the structureactivity relationship of the Rh complexes against NAE activity in vitro. Furthermore, complex 1 inhibited downstream CRLregulated substrate degradation and NF-kB signaling in cellulo, and was also found to exhibit prominent anti-proliferative activity against a human cancer cell line. Molecular modeling analysis revealed that 1 occupied the same binding pocket as MLN4924, the most potent NAE inhibitor to date. NAE promotes ubiquitination and the subsequent degradation of a BKM120 subset of proteins regulated by CRLs such as IkBaand p27. The IkBa protein plays a central role in repressing the activity of the transcription factor NF-kB, which is involved in important cellular processes including the immune response, programmed cell death, as well as cancer initiation and progression. The inhibition of NAE is therefore a potential approach to block the degradation of IkBa thus preventing NF-kB activation. On the other hand, the p27 is a cell cycle regulator which controls cell cycle progression during the G1 phase. The loss of function in p27 can lead to uncontrolled cell proliferation and the development of cancer. Inhibition of NAE activity in Caco-2 cells by 1 should be expected to result in the down-regulation of the CRL activity thus leading to the accumulation of CRL substrates. To examine this, we first investigated the ability of 1 to inhibit NAE-regulated IkBa degradation. Caco-2 cells were stimulated with TNF-a to induce IkBa protein degradation, which was monitored using Western blot analysis. Encouragingly, we observed that the induction of IkBa protein degradation by TNF-a was blocked by 1 in a dose-dependent manner, with potency comparable to the control compound  MLN4924 at 2.5 mM.
MLN4924 at 2.5 mM.
Treatment of cells with led to a block in internalization of transferrin receptor compared to untreated cellsor
Cells treated with the negative control of pitstop 2 that was provided by the manufacturer. However, internalization of MHCI was also inhibited. Although endocytosis of MHCI was inhibited by Pitstop 2, the antibody was still capable of binding to the surface of cells as shown by imaging the total cell-associated fluorescencein control and Pitstop 2 treated cells. Endocytosis of other CIE cargo proteins was examined in the (+)-JQ1 presence of pitstop 2. Internalization of CD59, a GPI-anchored protein with a trafficking itinerary similar to MHCI, was also blocked by pitstop 2. Three additional cargo proteins, which enter cells by CIE but take an alternative itinerary from that of MHCI and CD59 once inside the cell, were also examined. Treatment with pitstop 2  blocked endocytosis of these proteins, while in untreated cells, endocytosed CD44, CD98 and CD147 were observed in tubular recycling endosomes, as previously observed. The block in endocytosis induced by pitstop 2 was observed at shorter timesof internalization and could be reversed after 1 h of drug removal. The potent activity of pitstop 2 in blocking CIE was unexpected so we monitored its activity towards inhibiting transferrin and MHCI internalization with increasing doses of the compound. In HeLa cells we found that endocytosis of MHCI appeared to be somewhat more sensitive to the action of pitstop 2 than that of transferrin. We also noticed that even at high doses of pitstop, some transferrin still enters cells. Quantification of internalization of transferrin and MHCI revealed a shift in the dose-response curve with half-maximal inhibition for MHCI at around 6 mM and for transferrin around 18 mM. To further demonstrate that pitstop 2 blocks endocytosis of CIE cargo proteins, we turned to using a SNAP-tagged protein to quantify internalization in living cells. We recently developed a modification of labeling SNAP-tagged cell surface proteins using a releasable fluorescent tag on the benzylguanineligand. We transfected HeLa cells with a chimeric cargo protein consisting of the SNAP proteinattached to the extracellular portion of Tac, the IL2 receptor a-subunit. Tac enters cells by CIE and follows an intracellular itinerary similar to that of MHCI. Cells expressing SNAP-Tac were labeled with MG132 BG-S-S-594 and allowed to internalize for 30 min in the absence and presence of pitstop 2. Cells were then imaged live and fluorescence quantified prior toand then 1 min afteraddition of a cellimpermeable reducing agentthat cleaves the 594 label from the surface. This method allows for cell-by-cell quanitification of endocytosis. Pitstop 2 treatment reduced internalization of SNAP-Tac as compared to DMSO controls. The individual amounts internalized for each cell measured are plotted in Fig. 3B and clearly show a block in endocytosis in pitstop-treated cells. Furthermore, a similar amount of surface labeling with BG-S-S-594 was observed in control and pitstop-treated cells, indicating that pitstop did not interfere with BG binding to SNAP-Tac. Next, we examined the effect of pitstop 2 on internalization of transferrin and MHCI in two other human cell lines. In both BEAS-2B, a lung epithelial cell line, and in COS-7 cellsinhibition by pitstop of transferrin and MHCI internalization was also observed. We did note, however, that in these cell lines, endocytosis of both transferrin and MHCI appeared to be blocked by pitstop 2 with similar potencies. The shift in the dose-response curve observed in HeLa cells suggests that CIE may be more sensitive to the drug than CDE, raising the possibility that pitstop 2 has additional cellular targets besides the clathrin N-terminal domain.
blocked endocytosis of these proteins, while in untreated cells, endocytosed CD44, CD98 and CD147 were observed in tubular recycling endosomes, as previously observed. The block in endocytosis induced by pitstop 2 was observed at shorter timesof internalization and could be reversed after 1 h of drug removal. The potent activity of pitstop 2 in blocking CIE was unexpected so we monitored its activity towards inhibiting transferrin and MHCI internalization with increasing doses of the compound. In HeLa cells we found that endocytosis of MHCI appeared to be somewhat more sensitive to the action of pitstop 2 than that of transferrin. We also noticed that even at high doses of pitstop, some transferrin still enters cells. Quantification of internalization of transferrin and MHCI revealed a shift in the dose-response curve with half-maximal inhibition for MHCI at around 6 mM and for transferrin around 18 mM. To further demonstrate that pitstop 2 blocks endocytosis of CIE cargo proteins, we turned to using a SNAP-tagged protein to quantify internalization in living cells. We recently developed a modification of labeling SNAP-tagged cell surface proteins using a releasable fluorescent tag on the benzylguanineligand. We transfected HeLa cells with a chimeric cargo protein consisting of the SNAP proteinattached to the extracellular portion of Tac, the IL2 receptor a-subunit. Tac enters cells by CIE and follows an intracellular itinerary similar to that of MHCI. Cells expressing SNAP-Tac were labeled with MG132 BG-S-S-594 and allowed to internalize for 30 min in the absence and presence of pitstop 2. Cells were then imaged live and fluorescence quantified prior toand then 1 min afteraddition of a cellimpermeable reducing agentthat cleaves the 594 label from the surface. This method allows for cell-by-cell quanitification of endocytosis. Pitstop 2 treatment reduced internalization of SNAP-Tac as compared to DMSO controls. The individual amounts internalized for each cell measured are plotted in Fig. 3B and clearly show a block in endocytosis in pitstop-treated cells. Furthermore, a similar amount of surface labeling with BG-S-S-594 was observed in control and pitstop-treated cells, indicating that pitstop did not interfere with BG binding to SNAP-Tac. Next, we examined the effect of pitstop 2 on internalization of transferrin and MHCI in two other human cell lines. In both BEAS-2B, a lung epithelial cell line, and in COS-7 cellsinhibition by pitstop of transferrin and MHCI internalization was also observed. We did note, however, that in these cell lines, endocytosis of both transferrin and MHCI appeared to be blocked by pitstop 2 with similar potencies. The shift in the dose-response curve observed in HeLa cells suggests that CIE may be more sensitive to the drug than CDE, raising the possibility that pitstop 2 has additional cellular targets besides the clathrin N-terminal domain.