The second protocol concerns the determination of accuracy for positioning ligands in proteins active sites. This protocol was used to compare the two docking programs, SOL and the standard AutoDock 3.05. The first protocol showed a good to excellent quality in the SOL program for the selection of active inhibitors for four different target-enzymes from a large set of active and inactive ligands. The accuracy of CT99021 ligand positioning in the active sites of enzymes was defined by the root mean square deviation between ligand docked poses and experimental ligand poses taken from the Protein Data Bank. The grid of potentials representing thrombin-ligand interactions was calculated separately using the SOL_GRID program, before the initiation of the docking procedure. Throughout the docking studies, all ligands were considered fully flexible �C i.e., all topologically available torsional degrees of freedom were unfrozen and allowed to VE-821 rotate freely, directed only by ligand internal energy preferences in the frame of MMFF94. Bond lengths and valence angles were frozen in the course of the docking procedure. A fast decrease of preformed thrombin activity rises is vital in acute situations. Thus, it is reasonable in such cases to intravenously administer direct thrombin inhibitors to block hypercoagulation as quickly as possible. Our aim was to 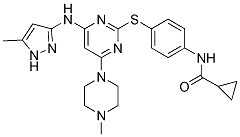 design new thrombin inhibitors for intravenous administration, whereby inhibitors can get directly to blood plasma where thrombin works. Thus, bioavailability was not an issue, and we were not restricted to ligands with low basicity in their P1 fragments. We have shown before that moderate plasma dilution in vitro with different artificial PSS produced hypercoagulation changes in the coagulation system. This fact suggests that plasma dilution, especially by crystalloid PSS, could also be a risk factor for the induction of thrombotic states during moderate hemodilution in vivo. The development of hypercoagulation has been shown to correlate with the infusion of large volumes of crystalloid solutions in patients. At present, the mechanism of this phenomenon is not clear; however, many investigators propose that during moderate hemodilution, the coagulation system is more sensitive to decreasing concentrations of coagulation inhibitors than to dilution of procoagulant factor precursors that are present in the blood in abundance. To prevent the development of hemodilutional hypercoagulation, we supplemented a crystalloid PSS with DTI. It was shown that the natural thrombin inhibitor antithrombin III could be used for this purpose. However, this inhibitor is isolated from human plasma and is thus very expensive and not completely safe with regard to the transmission of viral infections. Small molecule synthetic thrombin inhibitors are more suitable for this purpose. To be used in PSS, these inhibitors should be not only highly effective and safe, but also stable in aqueous solutions. The development of this kind of inhibitor was one of the objectives of our study. A majority of successful thrombin inhibitors have positively charged or neutral but easy polarizable P1 fragments. During thrombin-inhibitor complex formation, the P1 moiety of the inhibitor is located in the thrombin active site within a narrow cavity, exposing the carboxyl side chain of the Asp189 residue on its bottom. The severe spatial restrictions dictate the small size and hydrophobic nature of the P2 inhibitor position. In contrast, the restrictions on the P3 site are not as stringent because the corresponding binding site in the thrombin molecule is broad and exposed to the solvent.
design new thrombin inhibitors for intravenous administration, whereby inhibitors can get directly to blood plasma where thrombin works. Thus, bioavailability was not an issue, and we were not restricted to ligands with low basicity in their P1 fragments. We have shown before that moderate plasma dilution in vitro with different artificial PSS produced hypercoagulation changes in the coagulation system. This fact suggests that plasma dilution, especially by crystalloid PSS, could also be a risk factor for the induction of thrombotic states during moderate hemodilution in vivo. The development of hypercoagulation has been shown to correlate with the infusion of large volumes of crystalloid solutions in patients. At present, the mechanism of this phenomenon is not clear; however, many investigators propose that during moderate hemodilution, the coagulation system is more sensitive to decreasing concentrations of coagulation inhibitors than to dilution of procoagulant factor precursors that are present in the blood in abundance. To prevent the development of hemodilutional hypercoagulation, we supplemented a crystalloid PSS with DTI. It was shown that the natural thrombin inhibitor antithrombin III could be used for this purpose. However, this inhibitor is isolated from human plasma and is thus very expensive and not completely safe with regard to the transmission of viral infections. Small molecule synthetic thrombin inhibitors are more suitable for this purpose. To be used in PSS, these inhibitors should be not only highly effective and safe, but also stable in aqueous solutions. The development of this kind of inhibitor was one of the objectives of our study. A majority of successful thrombin inhibitors have positively charged or neutral but easy polarizable P1 fragments. During thrombin-inhibitor complex formation, the P1 moiety of the inhibitor is located in the thrombin active site within a narrow cavity, exposing the carboxyl side chain of the Asp189 residue on its bottom. The severe spatial restrictions dictate the small size and hydrophobic nature of the P2 inhibitor position. In contrast, the restrictions on the P3 site are not as stringent because the corresponding binding site in the thrombin molecule is broad and exposed to the solvent.
All posts by NaturalProductLibrary
Similarly conducted with the same protocol and in a close similar healthcare setting with important implications
In the United States, more than 400,000 hospital admissions per year for upper GI bleeding are estimated to occur, and mortality ranges between 3% and 14%; this has not changed in the past 10 years and increases with increasing age. Known risk factors for peptic ulcer bleeding are non-steroidal anti-inflammatory drugs use and Helicobacter pylori infection. More recently, selective serotonin reuptake inhibitors have been identified as another risk factor, and since then, 15 additional studies �Cincluding this one�C addressing this topic have been carried out. Albeit four studies found a strong significant association between SSRIs and upper GI bleeding, others found no association at all; thereby, the association remains a matter of controversy. The widespread use of antidepressants, particularly SSRIs, makes even small risks account for a large number of cases, converting this problem into an important public health issue. This fact, along with the lack of consistency of the findings in the studies carried out so far, has aroused a great interest on this subject. Our study has been conducted with carefully collected information to further understand the relationship between SSRIs and upper GI bleeding in the framework of a Perifosine clinical trial general study on risk factors of upper GI bleeding. In the construction of the model, patients were taken as level one, strata as level two, and hospitals as level three. In the estimation of the models we used the lmer function, implemented in the context of the lme4 R package. This function performs the fit by using the Laplace approximation, and the correlation structure within the random effects has only an additional constraint: the variance-covariance matrix must be symmetric and positive semidefinite. To construct these models, the absence or lowest level of exposure was taken as the reference category. We performed a bivariate analysis, using the CX-4945 variables of exposure and potential confounders; and a multivariate analysis, including those independent variables which yielded a statistical significance of less than 0.2 in the bivariate analysis. The independent variables with the highest level of statistical significance were successively eliminated from the original model, provided that the coefficients of the principal variables of exposure changed by no more than 10% and Schwarz��s Bayesian Information Criterion. In addition to each antidepressant group, the following variables were included in the model: alcohol and caffeine consumption, past history of GI disorders, family history of GI bleeding, osteoarthritis, number of medicines taken and the use of NSAIDs, salicylates, proton pump inhibitors, H2 antihistamines,  antacids, antiplatelet agents and anticoagulants. The protocol of the study was approved by the corresponding Ethics Committees of the participating hospitals. All patients were asked for an informed consent and, to be included in the study, they had to give a written informed consent. When our study protocol was first prepared, four studies had been published; three of those studies found odds ratios of 3 or more and only one, clearly heterogeneous in design, found no risk; thus, our study had power to detect an odds ratio of 2 and failed to detect an association between SSRI exposure and occurrence of upper GI bleeding of this magnitude; in it, we have found a clear association with NSAIDs in the range of that reported in the literature. When considering the different groupings of antidepressants there were no differences.
antacids, antiplatelet agents and anticoagulants. The protocol of the study was approved by the corresponding Ethics Committees of the participating hospitals. All patients were asked for an informed consent and, to be included in the study, they had to give a written informed consent. When our study protocol was first prepared, four studies had been published; three of those studies found odds ratios of 3 or more and only one, clearly heterogeneous in design, found no risk; thus, our study had power to detect an odds ratio of 2 and failed to detect an association between SSRI exposure and occurrence of upper GI bleeding of this magnitude; in it, we have found a clear association with NSAIDs in the range of that reported in the literature. When considering the different groupings of antidepressants there were no differences.
Used to further analyze the general properties of protein interfaces with a known inhibitor
The 2P2IDB contains a total of 17 protein/protein complexes corresponding to 14 families and 56 small molecule inhibitors bound to the corresponding target. There are a limited number of targets in the 2P2I database at this stage due to the structural prerequisites that were used. However, it is inevitable that high throughput structural genomic programs will generate a high  level of data. In addition, the development of improved methodologies for the development of small molecule inhibitors will rapidly lead to the discovery and structural characterization of disruptors of new PPI families. These new targets and their corresponding ligands will be incorporated into the database as they appear in the literature and the Protein Data Bank. To assess the characteristics of druggable PPIs, the general properties of the interfaces found in 2P2IDB were compared to those of representative datasets of heterodimeric complexes retrieved from BIBW2992 Bahadur and Zacharias and from the ProtorP server. Trauma to the CNS can BYL719 result in major disruptions in white matter tracts, including breakdown of myelin sheaths. Products of this myelin breakdown come in contact with the surfaces of severed axons and inhibit regeneration. The three known major myelin-derived inhibitors are Nogo-A, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein. All three bind with high affinity to the Nogo-66 receptor on axonal surfaces. Enzymatic cleavage of NgR confirms this effect, in that it increases axon regeneration. It was recently shown that phosphorylation of NgR by casein kinase II also inhibits binding of the myelin-associated proteins and promotes regeneration. Because NgR is a GPI-linked receptor and lacks an intracellular signaling domain, it relies on the transmembrane co-receptor, p75, to transduce the inhibitory signal. The final step in the signaling pathway is the activation of RhoA, a small GTPase that regulates actin polymerization and inhibits axonal elongation in its active form. Nogo-A, MAG, and OMgp activate RhoA through the NgR/p75 receptor complex, and this NgR/p75-complex/RhoA pathway is postulated to be responsible for the inhibitory signals that prevent axon regeneration. Recent pharmacological methods to overcome CNS myelin inhibition involved the use of an anti-Nogo antibody, RhoA inhibitors, a NgR antagonist peptide, and soluble NgR. There are potential problems with these inhibitors as therapeutic agents. For example, the direct blockade of RhoA with an inhibitor may disrupt other, crucial Rho-related cellular activities. In contrast, the anti-Nogo antibodies are only specific for Nogo and do not disrupt MAG or OMgp action. Because of this, it may be useful to identify high affinity inhibitors that more generally interact with the surface of NgR. Aptamers are single-stranded oligonucleotides that fold into unique three-dimensional structures, allowing them to bind to protein targets with high affinity and specificity. They are an alternative to therapeutic antibodies but can be chemically synthesized in a cell-free system. Furthermore, aptamers have a number of advantages over peptide and protein antagonists, including their relatively low cost of production, ease of GMP manufacture, and the simplicity with which they can be modified for stability, signaling, and immobilization. Studies have shown that aptamers have no or low immunogenicity, and are generally non-toxic, which is a great advantage in comparison to antibodies given the length of treatment period required for spinal cord injuries.
level of data. In addition, the development of improved methodologies for the development of small molecule inhibitors will rapidly lead to the discovery and structural characterization of disruptors of new PPI families. These new targets and their corresponding ligands will be incorporated into the database as they appear in the literature and the Protein Data Bank. To assess the characteristics of druggable PPIs, the general properties of the interfaces found in 2P2IDB were compared to those of representative datasets of heterodimeric complexes retrieved from BIBW2992 Bahadur and Zacharias and from the ProtorP server. Trauma to the CNS can BYL719 result in major disruptions in white matter tracts, including breakdown of myelin sheaths. Products of this myelin breakdown come in contact with the surfaces of severed axons and inhibit regeneration. The three known major myelin-derived inhibitors are Nogo-A, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein. All three bind with high affinity to the Nogo-66 receptor on axonal surfaces. Enzymatic cleavage of NgR confirms this effect, in that it increases axon regeneration. It was recently shown that phosphorylation of NgR by casein kinase II also inhibits binding of the myelin-associated proteins and promotes regeneration. Because NgR is a GPI-linked receptor and lacks an intracellular signaling domain, it relies on the transmembrane co-receptor, p75, to transduce the inhibitory signal. The final step in the signaling pathway is the activation of RhoA, a small GTPase that regulates actin polymerization and inhibits axonal elongation in its active form. Nogo-A, MAG, and OMgp activate RhoA through the NgR/p75 receptor complex, and this NgR/p75-complex/RhoA pathway is postulated to be responsible for the inhibitory signals that prevent axon regeneration. Recent pharmacological methods to overcome CNS myelin inhibition involved the use of an anti-Nogo antibody, RhoA inhibitors, a NgR antagonist peptide, and soluble NgR. There are potential problems with these inhibitors as therapeutic agents. For example, the direct blockade of RhoA with an inhibitor may disrupt other, crucial Rho-related cellular activities. In contrast, the anti-Nogo antibodies are only specific for Nogo and do not disrupt MAG or OMgp action. Because of this, it may be useful to identify high affinity inhibitors that more generally interact with the surface of NgR. Aptamers are single-stranded oligonucleotides that fold into unique three-dimensional structures, allowing them to bind to protein targets with high affinity and specificity. They are an alternative to therapeutic antibodies but can be chemically synthesized in a cell-free system. Furthermore, aptamers have a number of advantages over peptide and protein antagonists, including their relatively low cost of production, ease of GMP manufacture, and the simplicity with which they can be modified for stability, signaling, and immobilization. Studies have shown that aptamers have no or low immunogenicity, and are generally non-toxic, which is a great advantage in comparison to antibodies given the length of treatment period required for spinal cord injuries.
Regulators of carbohydrate metabolism predictive of benefits of DGAT1 inhibition combinations may find applications in spectrum of hematological malignancies
Interestingly, as opposite to what was observed in leukemia cells, HDAC and sirtuin inhibitors were poorly active and failed to show any cooperation in CD34 hematopoietic progenitors and in PBMCs. For classical HDAC inhibitors, preferential activity against malignant tissues has been reported. The fact that cancer cells frequently express higher amounts of certain HDACs, and a peculiar composition of the HDAC complexes in malignant cells have both been proposed as possible reasons for this selectivity. In contrast to Audrito and co-workers, we failed to detect increased SIRT1 expression in B-CLL cells as compared to healthy leukocytes. This  could be due to the fact that these authors compared B-CLL cells to healthy B cells, while in our case SIRT1 expression in B-CLL cells was compared to its levels in PBMCs. However, as a possible explanation for the preferential activity of combined sirtuin and HDAC inhibitors in leukemias, we found that HDAC inhibition increases Bax��s levels in leukemia cells, but not in healthy leukocytes. Thus, it is likely that, by removing one arm of the two-pronged mechanism that we found underlie this form of synergy, the cooperation between the two types of agents is disabled. Further studies should address the specificity of sirtuin and HDAC inhibitors for leukemic cells. However, regardless of the underlying mechanism, these data highlight a specific requirement for sustained sirtuin and HDAC activity by leukemia cells and suggest a possible Achilles�� heel of leukemias that could be exploited therapeutically. In conclusion, sirtuin inhibitors and HDAC inhibitors cooperate in turning off cellular mechanisms that protect leukemia cells from apoptosis. Co-administration of sirtuin and HDAC inhibitors should be further examined for clinical applications. Most biological processes are NVP-BEZ235 regulated by reversible phosphorylation, and kinases play a central role in signal transmission. Kinases interconnect different signalling pathways in time and space, and confer flexibility to the regulation and coordination of multiple biological processes including cell division, GW786034 moa apoptosis and survival among others. Furthermore, alteration in kinase function is a common underlying process to many pathological situations including cancer, inflammation, and neurodegeneration. The elucidation of the human kinome has opened up new possibilities to characterize and develop strategies to manipulate these regulatory processes with therapeutic aims. Kinase domains are very suitable for development of specific inhibitors, some of which have already been applied in cancer treatment, both for tyrosine kinases, such as PDGF/kit with imatinib in a variety of tumours, or to Ser-Thr kinases such as for B-Raf in melanomas. Kinase domains in an inactive state are more structurally diverse than their activated form. However, the main problem in development of specific inhibitors resides in the high conservation of the catalytic domain, which reduces the specificity of most inhibitors by targeting several kinases simultaneously, which makes them non specific. This crossinhibition results in a significant promiscuity, which can be the cause of unexpected side effects in clinical use. The inhibition promiscuity of a kinase can be predicted based on the conservation of specific residues within the kinase fold. The VRK kinase family received its name from vaccinia virus B1R, its unique kinase required for viral replication. The VRK family has a unique ortholog in C. elegans and D. Melanogaster, but is composed of three proteins in mammals.
could be due to the fact that these authors compared B-CLL cells to healthy B cells, while in our case SIRT1 expression in B-CLL cells was compared to its levels in PBMCs. However, as a possible explanation for the preferential activity of combined sirtuin and HDAC inhibitors in leukemias, we found that HDAC inhibition increases Bax��s levels in leukemia cells, but not in healthy leukocytes. Thus, it is likely that, by removing one arm of the two-pronged mechanism that we found underlie this form of synergy, the cooperation between the two types of agents is disabled. Further studies should address the specificity of sirtuin and HDAC inhibitors for leukemic cells. However, regardless of the underlying mechanism, these data highlight a specific requirement for sustained sirtuin and HDAC activity by leukemia cells and suggest a possible Achilles�� heel of leukemias that could be exploited therapeutically. In conclusion, sirtuin inhibitors and HDAC inhibitors cooperate in turning off cellular mechanisms that protect leukemia cells from apoptosis. Co-administration of sirtuin and HDAC inhibitors should be further examined for clinical applications. Most biological processes are NVP-BEZ235 regulated by reversible phosphorylation, and kinases play a central role in signal transmission. Kinases interconnect different signalling pathways in time and space, and confer flexibility to the regulation and coordination of multiple biological processes including cell division, GW786034 moa apoptosis and survival among others. Furthermore, alteration in kinase function is a common underlying process to many pathological situations including cancer, inflammation, and neurodegeneration. The elucidation of the human kinome has opened up new possibilities to characterize and develop strategies to manipulate these regulatory processes with therapeutic aims. Kinase domains are very suitable for development of specific inhibitors, some of which have already been applied in cancer treatment, both for tyrosine kinases, such as PDGF/kit with imatinib in a variety of tumours, or to Ser-Thr kinases such as for B-Raf in melanomas. Kinase domains in an inactive state are more structurally diverse than their activated form. However, the main problem in development of specific inhibitors resides in the high conservation of the catalytic domain, which reduces the specificity of most inhibitors by targeting several kinases simultaneously, which makes them non specific. This crossinhibition results in a significant promiscuity, which can be the cause of unexpected side effects in clinical use. The inhibition promiscuity of a kinase can be predicted based on the conservation of specific residues within the kinase fold. The VRK kinase family received its name from vaccinia virus B1R, its unique kinase required for viral replication. The VRK family has a unique ortholog in C. elegans and D. Melanogaster, but is composed of three proteins in mammals.
We have successfully identified two novel tetracycline-derived inhibitors of the propagation
The tetracycline-binding site of the TetR Selumetinib protein has been defined and the structure determined by crystallography. We found that the TetR protein shares similar characteristics with the E protein in the binding sites for the tetracycline derivatives. First, there is an appropriate volume in the binding sites. The volumes of the binding sites of various TetR crystals range from 359 A ? 3 to 495 A ? 3 whereas the BOG binding site on the E protein is 481 A ? 3, according to the tool program, QSiteFinder. Therefore, there is proper space for the tetracycline derivatives to fit into the BOG binding site. Second, there are hydrophobic surfaces in the pockets of both binding sites. Third, according to the results of a cross-docking test performed for TetR and the tetracycline derivatives, the binding sites of the DV E protein and TetR permit the binding of the tetracycline derivatives. In addition, the hydrogen bonds formed between the tetracycline derivatives and the DV E protein are similar to those between TetR and the tetracycline-derived ligands. Therefore, tetracycline derivatives should reasonably bind the BOG pocket of the DV E protein. On the other hand, only two of the derivatives are inhibitory; therefore, the atomic details of the functional ICI 182780 groups and the tetracyclic core must confer the inhibitory activity. Hence, we have analyzed the docked conformations and hydrogen bonding of the derivatives to assess the interaction between those compounds and the E protein. There are distinct differences between the effective and ineffective compounds; the effective compounds have their tetracyclic cores positioned inside the pocket while their side chains form hydrogen bonds with the residues located on the opposite sides of the wall around the pocket and are capable of creating steric hindrance to the conformational alteration of the E protein. In contrast, the ineffective compounds form hydrogen bonds only with one side of the wall and their cores lean away from the pocket. Next, on anatomiclevel, the predictedpositions of thetetracycline derivatives with the E protein are shown in Figures 6 and 7. The fused tetracyclic rings of rolitetracycline and doxytetracycline bind along the D9o strand and occupy the D9c space of the E protein. The residues 48�C52 are in the D segments. These compounds both interact mainly with Thr48, Glu49, Ala50, Gln200, and Gln271 through hydrogen bonds. Such a hydrogen-bonding network provides strong attraction forces to  stabilize the binding of rolitetracycline and doxytetracycline to the D9o strand and the kl b-hairpin. In contrast, although these compounds have the same tetracyclic core structures, neither tetracycline nor oxytetracycline is inhibitory. Both compounds form hydrogen-bonding networks with Thr48, Gln200, Gln271, Phe279, and Thr280; therefore, their tetracyclic rings are docked toward one side of the binding site and contact the surrounding hydrophobicresidues via van der Waals interactions, which are very different from those of rolitetracycline and doxytetracycline. During the process of E protein-host membrane fusion, the E protein structure is dramatically re-configured to allow the fusion peptide to properly interact with the host membrane. This event is marked by the rearrangement of the kl b-hairpin and the D9o segment in the BOG binding site. The docked positions of the inhibitors suggest that they occupy the D9c and kl b-hairpin spaces in the post-fusion state and form a stable hydrogen-bonding network. Therefore, these compounds block the rearrangement of the b-hairpin and D9o strand, and thereby block the rearrangement of domains II and I of the E protein during membrane fusion. Residues 48-52 are not only important to inhibitor binding but may also directly affect flavivirus membrane fusion. This hypothesis is consistent with previous reports that Gln52 may affect the pH threshold of fusion in flaviviruses. Our study has presented a cost-effective and time-saving screening process that is based on limited structural information.
stabilize the binding of rolitetracycline and doxytetracycline to the D9o strand and the kl b-hairpin. In contrast, although these compounds have the same tetracyclic core structures, neither tetracycline nor oxytetracycline is inhibitory. Both compounds form hydrogen-bonding networks with Thr48, Gln200, Gln271, Phe279, and Thr280; therefore, their tetracyclic rings are docked toward one side of the binding site and contact the surrounding hydrophobicresidues via van der Waals interactions, which are very different from those of rolitetracycline and doxytetracycline. During the process of E protein-host membrane fusion, the E protein structure is dramatically re-configured to allow the fusion peptide to properly interact with the host membrane. This event is marked by the rearrangement of the kl b-hairpin and the D9o segment in the BOG binding site. The docked positions of the inhibitors suggest that they occupy the D9c and kl b-hairpin spaces in the post-fusion state and form a stable hydrogen-bonding network. Therefore, these compounds block the rearrangement of the b-hairpin and D9o strand, and thereby block the rearrangement of domains II and I of the E protein during membrane fusion. Residues 48-52 are not only important to inhibitor binding but may also directly affect flavivirus membrane fusion. This hypothesis is consistent with previous reports that Gln52 may affect the pH threshold of fusion in flaviviruses. Our study has presented a cost-effective and time-saving screening process that is based on limited structural information.